
Cystic Fibrosis
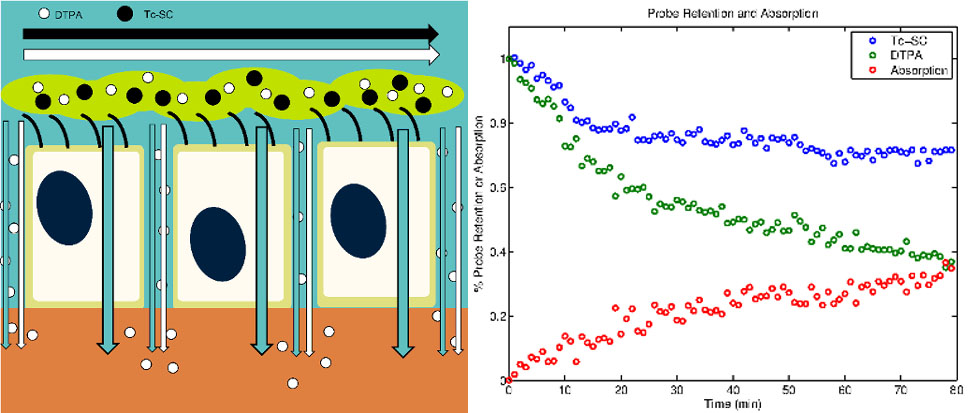
Cystic Fibrosis (CF) is a fatal autosomal disease originating from a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that causes decreased chloride secretion and dysregulation of sodium absorption in the airway epithelium, leading to dehydration of the airway surface liquid layer (ASL), mucociliary clearance (MCC) failure, chronic infections, inflammation, scarring, and respiratory failure. Recent therapeutic development in CF has focused on modulating ASL hydration through the correction of cell-level airway physiology. Therapeutic interventions for CF are studied in our lab on multiple levels. This includes in vitro studies human bronchial epithelial (HBE) and human nasal epithelial (HNE) cell cultures and functional imaging studies of MCC and ASL absorption in vivo. Since HNE cells can be donated through minimally invasive procedures these models could be used to predict personalized response for individual subjects. Prospective CF therapies are often identified by observing the change induced in electrogenic ion current in cultured cells. While these methods demonstrate that a bioelectric abnormality exists in patients with CF, they also require that the epithelium be flooded with a perfusion solution during the experiment. This may obscure the effect of therapies, as it is thought that ENaC and CFTR are volume-regulated. More representative conditions are those of “thin-film” cell culture experiments that our model is built to describe.
Using basic transport equations and data from multiple imaging studies of adult and pediatric CF patients and healthy controls we have developed a model describing the pathophysiological interactions between ASL absorption and MCC in different airway zones. We have also begun the development of an in vitro model that describes ion, liquid, and solute transport across the airway epithelium. Here we seek to more fully develop and specifically define key terms in the in vitro model and provide extensive data on in vitro therapeutic response. We further seek to integrate our in vitro and in vivo models to produce a multi-scale model that will allow for the prediction of therapeutic response in vivo based on measurements of response made in vitro.
People
Graduate students on the project include Florencio Serrano and Matthew Markovetz. Past and present undergraduates include Joey Nero, Jasmine Toney, Ryan Lacy, Lauren Musgrave (REU), and Nicholas Lotz (REU). The mentoring team includes Drs. Robert Parker, (mathematical modeling of biological systems), Timothy Corcoran (functional lung imaging), Carol Bertrand (epithelial biology), Joseph Pilewski (CF Pulmonology), and Michael Myerburg (CF cell cultures, CF Pulmonology).
Publications
Markovetz, Matthew R., et al. "A Physiologically-Motivated Compartment-Based Model of the Effect of Inhaled Hypertonic Saline on Mucociliary Clearance and Liquid Transport in Cystic Fibrosis." PLOS One 9.11 (2014): e111972. ( link)
Locke, Landon W., et al. "Quantitative imaging of airway liquid absorption in cystic fibrosis." European Respiratory Journal 44.3 (2014): 675-684. ( link)